4 靶向细胞周期蛋白, k0 |6 [& @3 i6 `0 m% B
细胞生长分裂必须依次经过准备阶段的间期(interphase)和有丝分裂期(mitosis)。间期(包括G1、S、G2期)的各项生命活动保证了M期分裂时所需的细胞内各成分的复制,每次有丝分裂的结束到下一次有丝分裂的结束构成一个完整的细胞周期。细胞周期的运行与否,受控于精密的细胞周期调控机制。该调控系统的核心是一组细胞周期依赖性蛋白激酶(Cyclin-dependent-kinase,CDKs),它们各自在细胞周期内特定时间被激活,通过磷酸化对应的底物,驱使细胞周期的完成。CDKs的时相性激活依赖于时相表达的周期素(cyclin)以及周期素依赖性激酶抑制剂(cyclin-dependent kinase inhibitors,CKI)控制。另外,除了这种正常生理条件下的周期进程调控,在长期的进化过程中,细胞建立了一套保证细胞周期中遗传信息的完整性和准确性的检查机制,即细胞周期检查点(checkpoint)。当细胞周期进程中出现异常事件,如DNA损伤或DNA复制受阻时,这类调节机制就被激活,及时中断细胞周期的运行。待DNA修复或排除了故障后,细胞周期才能恢复运转。
6 j: ?7 z2 Z0 A/ B8 n6 w
* Z4 s6 m4 J2 F; J' [3 K, k5 I 在细胞的癌变过程中,通常都伴随着cyclin的过度表达和CKIs的缺失,CDK的活性失去控制,细胞周期处于失控状态;肿瘤细胞的另外一个特点是细胞周期检查点缺陷,造成对细胞损伤应答的缺失。然而,这种周期检查点关卡的缺失,使得细胞对外界的损伤更加敏感又能被应用于肿瘤的治疗,增加放化疗的敏感性。基于肿瘤细胞的上述特点,恢复肿瘤细胞的周期调控和取消检查点等都成为潜在的抗肿瘤作用靶点。具体策略包括对CDK的直接催化抑制,阻碍CDK的激活,干扰周期素与CDK的相互作用,影响周期素水解失活和抑制细胞周期检测点等。目前,已经有多个细胞周期的调节剂进入了临床研究,其中植物来源的黄酮类物质flavo-piridol能明显抑制CDK1、CDK2和CDK4,阻碍细胞通过G1/S和G2/M期检测点,能抑制多种肿瘤细胞的生长,已经处于临床Ⅱ期研究。另外,星型苞菌素(Stauros-porine)的类似物UCN-01除了抑制PKC外,还可直接抑制CDK1和CDK2的活性和细胞周期检测点激酶chk1的活化,目前正在美国和日本进行临床Ⅱ期实验。还有Paullones类似物、嘌呤霉素类似物(Pruines)等都对不同的CDK分子显示出抑制作用。 l7 k" ~9 C/ L% ^* `8 O# z
4 G$ B- o- Q7 u* W5 组蛋白去乙酰酶抑制剂(histone deacetylase inhibitor,HDACi)
( j9 D5 \( K/ C 肿瘤的发生和诸多基因特别是癌基因的异常表达密切相关,而染色体结构是调控基因表达的重要因素。通常情况下,凝缩的染色体会抑制基因的转录,而有转录活性的基因一般位于松散的染色体区域。染色体的基本单位核小体是由组蛋白(Histone)和DNA组成的,其中组蛋白的转录后修饰,包括乙酰化、磷酸化和甲基化能够改变核小体的高级结构,进而影响着染色体的高级结构和基因的转录调控。细胞内一对功能相互拮抗的蛋白酶,组蛋白乙酰基转移酶(Histone acetyl-transferases,HATs)和组蛋白去乙酰酶(Histone deace-tylases,HDACs)共同决定着组蛋白的乙酰化和去乙酰化。HAT可乙酰化组蛋白末端碱性氨基酸的氨基,使核小体舒展,激活基因转录。而HDAC与之功能相反,抑制基因转录。
* N) B+ y3 M# Q# G0 J! `6 s* N
" E9 h/ V# G% d6 ] 近些年的研究发现HDAC作为调控基因转录的关键蛋白酶,其功能异常与肿瘤的发生和发展有直接关系。当HDAC过度表达并被转录因子募集时,会抑制某些基因的正常表达。这种因HDAC活性过高引起的异常转录抑制在肿瘤中非常普遍,因此HDAC成为抗肿瘤药物最具潜力的靶点之一。抑制HDAC的活性能引起组蛋白高度乙酰化,重新激活某些抑癌基因的转录并引起多项下游效应,包括促进肿瘤细胞分化、使肿瘤细胞阻滞于G1或G2期以及诱导肿瘤细胞凋亡,从而实现其抗肿瘤作用。另外,研究发现HDACi还能激活主要组织相容性复合物、细胞间粘附分子ICAM-1、干扰素Ⅰ/Ⅱ等分子的转录,促进免疫细胞的识别和激活。HDACi还能抑制缺氧诱导的VEGF表达,抑制新生血管生成。
' H/ Y n. W4 q2 _
" N* U/ k* s% i& K- Z 来自真菌的Trichostatin A(TSA)是发现的首个能高效抑制HDAC的羟胺类天然产物,但存在着天然含量低、体内代谢不稳定的缺点。目前已经有10多个不同结构类型的HDACi进入了Ⅰ/Ⅱ期临床试验,用于白血病和实体瘤的治疗。这些药物大多能在有效剂量显示出较好的耐受性,并显示出抗p-糖蛋白介导的多药耐药作用。& a8 e3 E# d- A( V% w; j' D! ~
+ T& M4 e* u6 M# e
6 靶向泛素-蛋白酶体通路(ubiquitin-proteasome system, UPS)+ a S" O$ h& V; T
蛋白降解调控是细胞信号转导的一个重要方面,与基因转录水平的调控相比,这种转录后调控还能保证细胞在遇到外界刺激时更加快速的做出反应。UPS是真核细胞内依赖ATP的非溶酶体蛋白质降解途径,负责调控细胞内多种蛋白的水解过程,其中包括许多调节细胞生长、信号转导、基因转录和凋亡的重要分子。泛素介导的蛋白降解是一个复杂的多级反应,其过程主要是利用泛素活化酶E1、泛素结合酶E2与泛素-蛋白连接酶E3,将泛素连接至目标蛋白作为标识,并送至20S蛋白酶体进行降解,最后由泛素分解酶将泛素分解并回收再利用。2 ~/ @$ W/ Y9 b% Y/ j- ~ n
8 M8 N* J. P! c- A1 y
由于UPS通路与肿瘤的发生、生长和转移都密切相关,该级联反应中各个环节都成为抗肿瘤药物作用的潜在靶点。例如,通过阻断泛素分子C末端的腺苷酸化或与ATP分子竞争结合的策略来阻碍泛素的激活;根据E1与E2相互作用的结合域特异地设计能够干扰其相互作用的小分子化合物阻碍泛素分子在E1和E2之间的传递等。其中特别值得一提的是靶向E3连接酶和其下游的蛋白酶体。
& p. K9 s9 f4 L, P5 L& T1 a$ O* y: z7 t3 Q
E1、E2和E3构成了金字塔形的级联放大系统,最下游的E3通过识别不同底物决定着整个泛素化过程的特异性。抑制泛素连接酶E3的功能可以通过抑制其与上游E2或下游蛋白底物的作用两条途径来实现。目前还没有发现针对E3和E2相互作用的抑制剂,但是对后者的研究已经取得了一些进展,其中最典型的例子就是MDM2对p53蛋白降解的调控。MDM2分子具有E3连接酶活性,通过泛素化调控p53的降解。Nutlins是首个发现的阻碍p53和MDM2相互作用的小分子抑制剂,其空间构象和p53分子中与MDM2作用的氨基酸残基非常相似,可以与p53分子竞争MDM2的结合位点。Nutlins在体内外都体现出抗肿瘤活性,对正常组织没有明显毒性。另外一个小分子抑制剂RITA早在1990年就发现其抗肿瘤活性,但在最近才发现其作用机理是与p53的N端结合,阻碍了MDM2对p53的识别,并稳定p53分子N端α-螺旋结构域。
/ y; F% ^# l1 X8 ~) e
2 v" ~6 d& b: G" b8 V1 w: s. X. ?% I E3连接酶下游的多个环节,包括蛋白酶体、参与泛素游离再循环的金属异肽酶(metal-loisopeptidase)以及对多聚泛素链的识别等都可能影响UPS通路。其中,首个上市的以UPS为靶点的小分子抑制剂bortezomib(Velcade,PS-341)就是直接抑制于蛋白酶体活性。9 `5 g1 V5 ^: M
% Z9 b2 R# A) }% ?* g) g+ G0 Z该化合物已先后于2003年5月和2004年4月被美国FDA和欧盟药品审评管理局(European Agency for the Evaluation of Medicinal Products ,EMEA)批准用于复发性和难治性多发性骨髓瘤的治疗。最近FDA又批准其作为一线药物用于已接受过至少一次治疗的多发性骨髓瘤患者。进一步在血液系统肿瘤、实体瘤以及非霍奇金淋巴瘤中的应用正在研究中。/ ? E' S6 Y9 `6 b4 f4 L
8 w( V# l, D& z: T7 靶向DNA损伤修复系统$ Q$ I+ a: {2 P. X
除了上述直接影响肿瘤细胞增殖调控的信号通路外,特异靶向DNA损伤修复通路中的一些关键分子也成为抗肿瘤药物研发的一个重要方向。很多传统的抗肿瘤药物包括烷化剂、DNA嵌入剂、拓扑异构酶抑制剂、抗代谢物等都是通过直接或间接造成不同形式的DNA损伤来实现其抗肿瘤作用。在外界损伤的刺激下,细胞能启动6条修复通路来分别应对不同类型的损伤:①直接修复(direct repair,DR)通路修复O6-烷基鸟嘌啉引起的损伤;②碱基切除修复(base excision repair,BER)针对因氧化还原或烷基化引起的碱基损伤;③核苷酸切除修复(nucleotide excision repair,NER)修复因辐射、化学药物或蛋白-DNA交联引起的核苷酸水平的损伤;④碱基错配修复(mismatch repair,MMR)纠正碱基错配;⑤同源重组修复(homologous repair,HR);⑥非同源的末端连接(non-homologous end-joining,NHEJ)通路,其中后两条通路专门修复DNA双链断裂(DNA double strand breaks,DSBs)。这些通路的激活往往削弱了化疗药的抗肿瘤效果,成为产生耐药的一个重要因素。' q% o0 A+ ^0 t& o, v9 Q7 G1 y4 d
, P9 `( `$ n: T5 a) v 目前,针对上述多条通路中一些关键分子的抑制剂研究都有不同程度的进展。如O6-苄基鸟嘌呤( O6-BG)及其多个衍生物能特异抑制DR通路中O6-烷基鸟嘌啉-DNA 烷基转移酶(AGT),目前正在作为耐药逆转剂与甲基化剂合用进行临床试验。还有靶向抑制BER通路中的多聚(ADP-核糖)聚合酶(poly[ADP-ribose]polymerase,PARP)的AG014699、NU1025、AG14361和INO-1001分别处于Ⅰ~Ⅲ期临床实验。这些抑制剂与烷化剂、拓扑异构酶I抑制剂、顺铂或γ-射线联合用于肿瘤细胞或者小鼠移植瘤,能分别增加后者引起的DNA损伤、细胞生长抑制和荷瘤小鼠的存活时间。现今,关注最多的是修复DSBs的HR和NHEJ通路,分别在这两条通路中扮演关键角色的ATM和DNA-PK都是PI3K激酶家族成员,前文已经提到的wortmannin、LY294002和咖啡因等PI3K家族的广谱抑制剂都能通过抑制这两条修复通路的活性起到放化疗增敏的作用。另外,还有ATP的竞争抑制剂KU-55933能显著抑制ATM活性,且作用明显强于对PI3K家族其它成员的抑制效果,能增强ATM高表达细胞株对离子辐射和VP16、阿霉素、喜树碱等造成DSBs药物的敏感性。DNA-PK的选择性抑制剂IC87361、Nu7026、Vanillin和SU117582也有相似的作用。; Q+ Y7 k" d. N9 [9 s: H
0 ~7 c. @ x( K5 p+ b
结语3 ?+ Z: H$ M5 M1 t; T# ]8 u
多个分子靶向药物的成功上市为新一代抗肿瘤药物的研发提供了乐观的前景。但是,必需看到靶向信号通路抑制剂只有在该信号通路高度激活的肿瘤上才产生更好地疗效,因此合理的临床设计是体现其疗效的必要前提。只有在合理的研究策略、全面的临床前评价和
4 m! C3 a) n2 X( k$ R% q" X/ j7 `/ b. |+ \4 L8 i
严谨合理的临床试验的共同配合下才能开发出更多有希望的靶点特异性抗肿瘤新药。 |
 汇总4月最新:临床试验招募| 小细胞
"化疗±免疫后进展了,还能怎么办?"——这是许多小细胞肺癌患者和家属的困境。
临
汇总4月最新:临床试验招募| 小细胞
"化疗±免疫后进展了,还能怎么办?"——这是许多小细胞肺癌患者和家属的困境。
临
 母亲肺腺癌,9年靶向耐药后,如何选
2017年5月,因持续干咳入院检查,肺部弥漫型结节,支气管镜确诊肺腺癌,四期。基因检
母亲肺腺癌,9年靶向耐药后,如何选
2017年5月,因持续干咳入院检查,肺部弥漫型结节,支气管镜确诊肺腺癌,四期。基因检
 新人求助
我爸爸身体不适 26 年 4 月 10 号去医院检查,初步结果是肺肿可能是肺癌,因年纪大 7
新人求助
我爸爸身体不适 26 年 4 月 10 号去医院检查,初步结果是肺肿可能是肺癌,因年纪大 7
 母亲肺腺癌晚期骨转移,EGFR21突变,
母亲1970年出生,56周岁,身高163cm,体重98kg(196斤)。2025年9月23日肺炎住院,胸
母亲肺腺癌晚期骨转移,EGFR21突变,
母亲1970年出生,56周岁,身高163cm,体重98kg(196斤)。2025年9月23日肺炎住院,胸
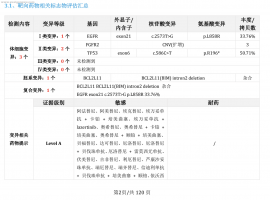 肺癌术后五年,脑膜转移治疗三年,伏
2025年8月14日更新: 终于输液港血培养结果为阴性了,14天美平消炎出院了,昨天开始双
肺癌术后五年,脑膜转移治疗三年,伏
2025年8月14日更新: 终于输液港血培养结果为阴性了,14天美平消炎出院了,昨天开始双






 + Y6 j+ x( \8 l9 Y" r' N
+ Y6 j+ x( \8 l9 Y" r' N 显身卡
显身卡


